Could disabling one protein cure the common cold?
Cold viruses don’t reproduce in mice and human cells lacking this protein
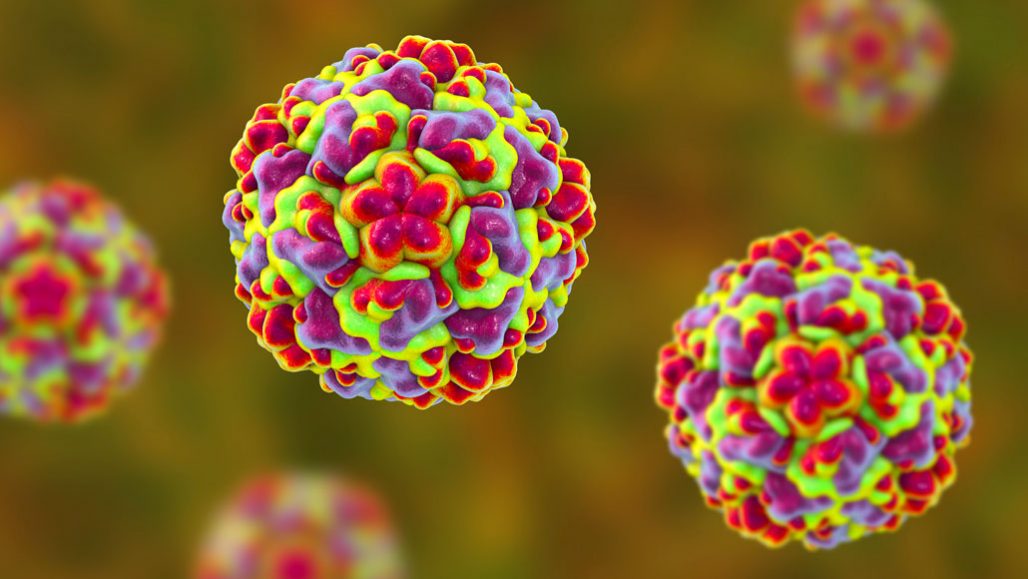
Rhinoviruses (shown in this drawing) are behind most cases of the common cold. New research suggests a way to stop them, and others, from spreading in people.
Dr_Microbe/iStock/Getty Images Plus
By Sofie Bates
Scientists may be one step closer to a cure for the common cold. They aren’t fighting the viruses that cause the infection. Instead they’re looking to make human cells — the hotel rooms in which viruses reproduce — an unwelcome place.
Viruses can’t reproduce on their own. They must hijack the machinery in cells to do that. So they temporarily infect those cells to make more of themselves. Then they explode open their host cell. This releases huge quantities of new infectious viruses.
Researchers have now identified a key protein that some viruses need to multiply inside human cells. Disabling that protein might stop a cold virus dead in its tracks. Or that’s the idea described September 16 in Nature Microbiology. Jan Carette is a microbiologist in California at the Stanford University School of Medicine. His team has just shown that in mice and human cells engineered to lack this protein, the viruses couldn’t replicate. No replication, no spread.
“It’s not quite a cure for the common cold,” says Ellen Foxman. She’s an immunologist at Yale School of Medicine in New Haven, Conn., who was not involved in the study. Still, she finds, the new work is “an interesting step forward.”
Colds are our most common infectious disease. On average, adults catch two or three each year. Children may get even more. Any one of a few hundred viruses, including rhinoviruses (RY-no-vy-russ-es), can cause these infections. That’s one thing that has made it so hard to find a cure for the cold. But there’s a second challenge, too. These viruses mutate — undergo spontaneous genetic changes — quickly. One or more of those changes may eventually leave them able to resist almost any drug thrown at them.
That’s why Carette’s group focused on the human host. The team wanted to see if it could identify the human proteins that many viruses use to replicate. Genes make a cell’s proteins. So these researchers hunted down the gene behind the protein that viruses take over.
Carette and his colleagues started by using a molecular scissor — the gene-editing tool CRISPR — to systematically snip out chunks of DNA from human cells growing in lab dishes. In the process, they built a library of altered cells, each one missing a single protein-making gene. The researchers then infected these altered cells with two types of viruses. One type causes colds. The other has been linked to nerve-related diseases.
Then the scientists used different viral proteins like hooks. They dipped them into the stuff inside human cells to see which proteins these viral hooks latched onto and pulled out. That let the team identify which human proteins were interacting with viral ones. This was a sign that the virus needed those human proteins to hijack a cell’s replicating machinery.
The viruses repeatedly fished out one particular protein. Known as SETD3, tests showed that the viruses needed it to take over a cell. Scientists knew this protein could affect actin proteins — ones that help muscles contract. That it might also aid viral infections came as a big surprise.
Tests show the anti-viral tactic has promise
To test the importance of SETD3, the researchers injected viruses into mice that had been engineered to lack a working version of that gene. And sure enough, no mice got sick. Human lung cells that also lacked the gene also remained healthy. (Lung cells are often used in these types of studies because they are especially susceptible to many cold-triggering rhinoviruses.)
Especially exciting: Running similar tests with other, potentially more serious, viruses suggested the approach may thwart more than just the common cold.
Engineered human cells didn’t become infected when they were exposed to viruses that cause hand, foot and mouth disease and a polio-like spinal-cord disease called acute flaccid myelitis (Flaa-sid My-eh-LY-tis). And when mice were exposed to these viruses, the rodents without a working SETD3 gene were much more likely to survive than those with a working gene.
“We have identified an excellent target,” says Carette, of SETD3. Still, it’s not clear whether disabling that gene and its protein might cause new problems. The engineered mice survived, appeared healthy and were fertile. They could not, however, push their pups out of the womb during birth. Says Carette, this might be due to the protein’s role in muscle contractions.
Scientists don’t fully understand what this gene does in people, so getting rid of it might have some nasty side-effects, notes Vincent Racaniello. He’s a virus expert at Columbia University in New York City who wasn’t involved in the work. “The authors show that mice lacking the gene for SETD3 are viable and resistant to infection. However,” he says, “this observation does not mean that SETD3 in humans is [not needed].”
Instead, the researchers think their best bet is to search for a drug that makes it seem like the gene isn’t working. The drug might block the human protein and its viral counterparts from interacting. Or, it might destroy the SETD3 protein — but only when it is interacting with the viruses.
Such drugs are still a long way off. “The question is always ‘When can I buy it over the counter?’” Carette says. The challenge, he notes: “Drug development takes time.”